Projects
 Our over-arching interest in the Klomp laboratory is to understand how specific protein kinases and their downstream targets contribute to cancer cell development, growth, maintenance, migration, and acquired resistance. By understanding these biological processes, we can more effectively develop protein kinase inhibitor-based therapies.
Our over-arching interest in the Klomp laboratory is to understand how specific protein kinases and their downstream targets contribute to cancer cell development, growth, maintenance, migration, and acquired resistance. By understanding these biological processes, we can more effectively develop protein kinase inhibitor-based therapies.
We currently are focused on studying ERK in Pancreatic ductal adenocarcinoma (PDAC), because of the nearly ubiquitous addiction of PDAC to oncogenic KRAS and the importance of ERK in driving that dependence. PDAC is the third leading cause of cancer deaths in the United States and is characterized by a 95% rate of mutational activation of the KRAS oncogene. ERK is a critical component for maintaining KRAS mutant PDAC growth as well as a major contributor of resistance to direct KRAS/RAS inhibitors in the clinic. Despite the highly successful development of potent and selective inhibitors of each node of the ERK MAPK cascade, when used as monotherapy, they have shown limited clinical efficacy against RAS-mutant cancers. Two key issues have contributed to this outcome, toxicity for normal tissues and de novo or treatment-induced acquired resistance in cancer cells. Research in the Klomp laboratory is focused on the delineation of the mechanisms by which ERK drives KRAS-dependent cancer growth to guide the development of more effective anti-ERK therapies.
- Project 1: ERK localization
- Project 2: ERK substrates
- Project 3: Functional evaluation of ERK regulated transcripts and phosphoproteins in PDAC
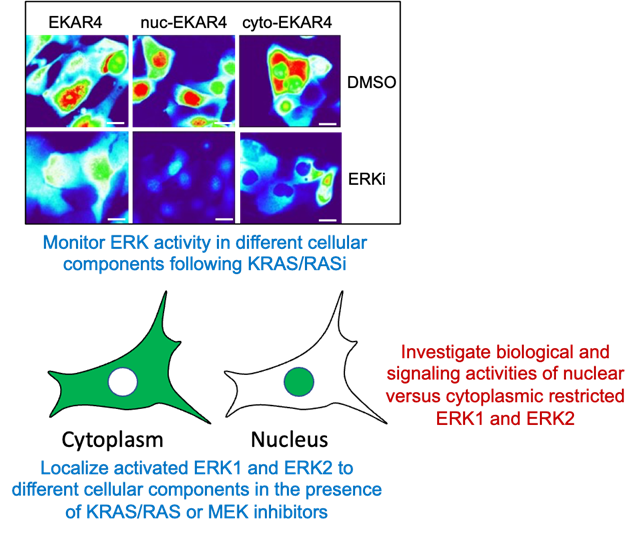
Objective: Determine the role of nuclear versus cytoplasmic ERK activity in the growth of KRAS-mutant cancer.
![]()
One major unresolved issue is how ERK activity in different subcellular compartments supports cancer growth. To address this, we are taking several complementary approaches to gain a better understanding of the role of cytoplasmic and nuclear ERK activity in supporting KRAS-dependent PDAC growth.
Recently, a variety of different KRAS and RAS inhibitors have become available and are currently being evaluated in the clinic. Using a panel of PDAC cell lines with varying degrees of sensitivities to inhibitors targeting the RAS-RAF-MEK-ERK pathway we are assessing the kinetics of nuclear versus cytoplasmic ERK activity using targeted ERK Kinase Activity Reporters (EKAR). We have shown previously that ERK is active throughout a PDAC cell and that PDAC cells rely on both ERK regulated phosphoproteins that are nuclear and cytoplasmic localized.
Using our recently described constitutively active ERK1 and ERK2 constructs we are determining what the contributions of nuclear versus cytoplasmic directed ERK activity are toward driving ERK mediated:
1) resistance mechanism to KRAS/RAS and MEK inhibitor growth suppression
2) signaling: transcriptome, phospho/proteome, and metabolome
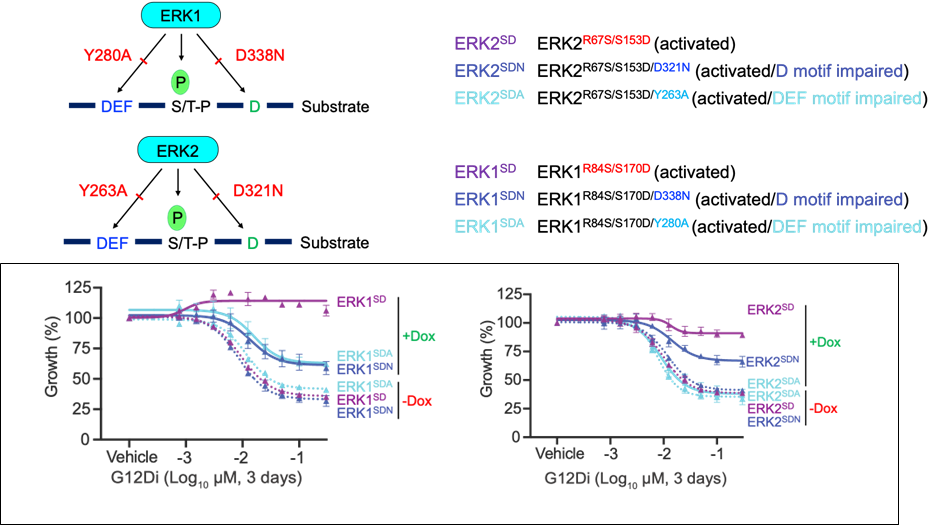
Objective: Determine the role of D versus DEF motif containing ERK targets in the growth of KRAS-mutant cancer.
![]()
ERK phosphorylates serine/threonine residues adjacent to a proline and direct ERK substrates usually possess either a D-site/D-(docking) motif and/or a F-site/DEF motif. We previously found that for ERK to rescue KRAS inhibitor growth suppression in PDAC cells it needed to be able to interact with both D- and DEF-motif containing substrates. Additionally, we found that D-motif containing substrates were localized throughout the cell and DEF-motif substrates were enriched in the nucleus.
Ongoing studies in the laboratory are evaluating the contribution of these two groups of ERK regulated phosphoproteins to ERK mediated signaling in KRAS mutant PDAC. Specifically, we are currently looking at the contribution to the ERK driven transcriptome, phospho/proteome, and metabolome.
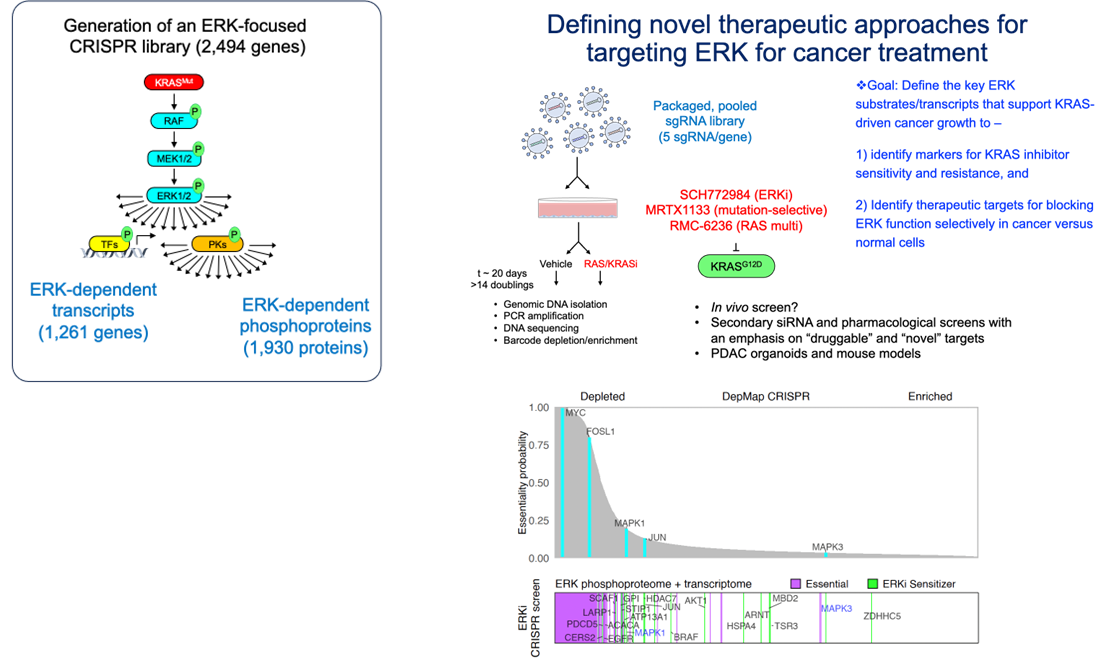
Objective: Identify ERK substrates that are synthetic lethal with KRAS/RAS and ERK inhibition.
![]()
We previously designed and generated a CRISPR-Cas9 genetic loss-of-function screen library, targeting ERK regulated phosphoproteins and/or transcripts based on our comprehensive ERK-dependent phosphoproteome/transcriptome studies in KRAS-mutant PDAC. We have and are continuing to perform screens, using this library to determine how ERK contributes to PDAC tumorigenesis as well as identify new ERK dependent targets to combine with KRAS/RAS inhibitors using this library.